| |
|
|
Documentation of HMMEditor 1.0 |
HMMEditor stands for profile Hidden Markov Model (pHMM) Visual Editor. It is a tool to visualize and edit pHMM in HMMer format. HMMer format is also used by Pfam protein database. Unique features of HMMEditor includes:
- Visualize pHMM in different views
- Edit profile HMM
- Show Viterbi path
-----------------------------------------------------------------------------------------
Visualize pHMM
Visualize pHMM is as easy as open a hmm file in HMMVE. Use the menu command "File"->"Open Hmm" to open a Hmmer format hmm file. You will see pHMM layout in Hmm Model panel right after that. Only pHMM in Hmmer format(Plan 7) is supported in HMMVE. HMMVE support Pfam database directly. You can download pfam database from ftp://selab.janelia.org/pub/Pfam/. The most popular database is Pfam-A.full.gz. Download it and unzip, you will a single large text file, which contains lots of pHMM in Hmmer format. You can use the menu command "File"->"Open Pfam" to open this pfam database file.
 |
Pfam Indexer |
Pfam indexer dialog shows all pHMM names and descriptions inside a pfam database. Since the indexing process may take more than one minute, so the indexer works in backgroud. All retrieved pHMM is shown in the table. The table keeps growing while indexer is working. You can select the desired pHMM without waiting for indexer complete. You can type in the phrase you want to search inside the "filter" text box. All pHMMs whose name or description contains that phrase will remain in the table, others are filtered out. Note that the search will ignore case, and you can put regular expressions into filter phrase. As before, you do not need to wait for the completion of the indexer to filter the result. However, There is one benefit if you wait for the competion of the indexer. Once indexing is finish, next time you open the same database, indexer can reuse the previous result and there is no need to index again. Once you find the desired pHMM, double click, or single click then press "Select" button, HMMVE will open that pHMM.
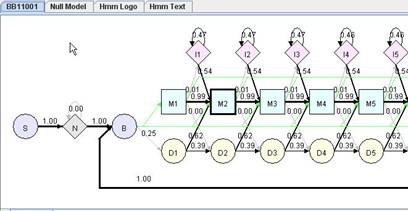 |
Hmm Model |
The layout of pHMM is described in Hmmer user guide. The whole profile HMM starts from start (S) state and ends at terminal (T) state. The core of HMM between beginning (B) and ending (E) states consists of the matching (M) states, insertion (I) states, and deletion (D) states. A matching state represents a fairly conserved position. Each matching state has a deletion state associated with it, allowing the deletion of the matching state (or position). Each matching state except for the last one also has an insertion state associated with it, allowing the insertion of additional positions after it. Transitions between I and D states are not allowed. N and C are two special states to accommodate additional insertions before and after the conserved regions of a family of sequences, which allows local alignment between a sequence and the HMM(i.e. matching a part of a sequence against the core of HMM between state B and state E). J state joins the end of a profile HMM to the beginning. J state allows aligning a sequence against the core of a profile HMM multiple times, which is called multi-domain alignment (domain duplication). Another interesting feature of the profile HMM is that there is a transition from B to each M state, and a transition from each M state directly to E state. These transitions make it possible to match only a part of the model against a sequence, allowing local alignment with respect to the HMM. Each M, I, N, C, J states has an emission probability vector derived from input sequences.
In generated Hmm Model view, the thickness of the transition line is proportional to the probability of the transition. The thickness of a border of an M state indicates the level of conservation.
When the mouse enter into an matching node, insertion node or other applicable node, emission probablity of that node will be shown in emission chart on the left bottom part of the HMMVE window.
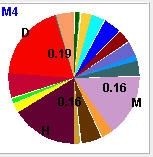 |
Emission Chart |
In Hmm Model view, you can zoom in/out the model using menu command. You can increase/decrease precision of transition probability on the model using menu command increase/decrease precision. You can choose to view the label of matching node as node name or consensus, or hide unnecessary connection (such as connection from B to M and the probability is 0). After dragging nodes around, you can use restore the nodes to default position using "default layout" command in menu.
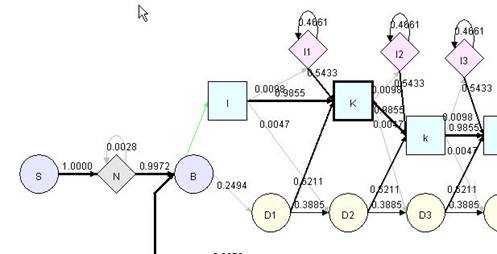 |
Hmm Model after numerous rearrangement |
You can save the Hmm model layout as it show in HMMVE window using menu "File"->"Save Image".
Each pHMM has a corresponding null model. Null Model is fully described in Hmmer user guide. Transition G->G control the length of the sequence generated. Emission probability of node G is the background occurrence frequencies of the 20 amino acids.
You can choose to view null model by clicking "Null Model" tab. In "Null Model", you can do similar operations as "Hmm Model" view, except that now you operate in a view consists of only two nodes.
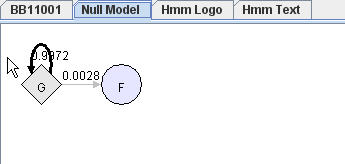 |
Null Model |
You can view Hmm Logo by clicking "Hmm Logo" tab. Hmm Logo is generated on demand, so it will take several seconds after you clicking "Hmm Logo" tab. The benefit is whenever you change Hmm Model in "Hmm Model" view or "Null Model" view, this change will be reflected in the generated Hmm Logo.
A HMM Logo consists of a serial of character stacks (column) separated by light red lines. Each stack represents a matching state. The lines separating neighboring stacks represent an insertion state. The height of the stack shows how significantly the emission probability of a matching state deviates from the background emission probability, i.e relative entropy (or information content). Internally, the height of each character is proportional to its information content. The width of each stack or line is determined by the hitting probability of its corresponding state.
In HMMVE, Hmm Logo is one view to visualize a pHMM. If generating Hmm Logo is your primary goal, please go to Hmm Log website directly. They generate better Logo than HMMVE.
All images in "Hmm Model", "Null Model" and "Hmm Logo" view can be save as image file. The saved image is just what you can see in HMMVE window after any rearrange and zoom operations.
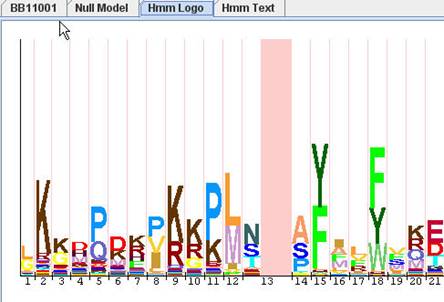 |
Hmm Logo |
You can view hmm text file by clicking "Hmm Text" tab without saving the model, then open the model file in a text viewer. Hmm text is generated on demand, so it always reflect the latest change in Hmm model. You can copy the text by selecting the text and press copy key of your operating system. Eg, in Windows, copy key is "Ctrl-C".
 |
Hmm Text |
Editing pHMM
You can edit transition and emission probability of applicable node in pHMM in "Hmm Model" view. You can also edit background emission frequency by editing in "Null Model" view.
To edit a node, simply right click an applicable node, choose "Modify Node". Alternatively, you can double click a node to invoke node modification dialog directly.
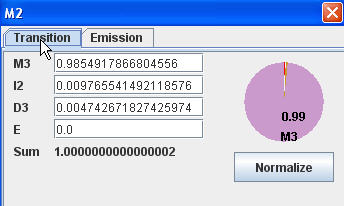 |
Modify transition probability |
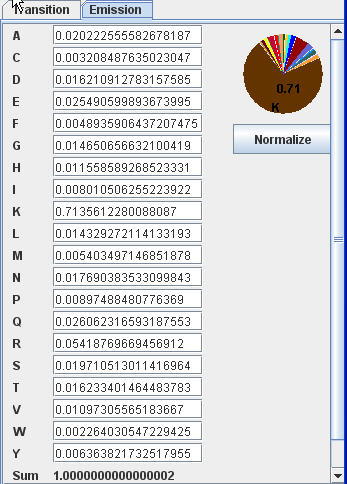
The transition probability of each node stands for the transitions origin from that node. The labels in "Modify Transition Probability" dialog show the transition destination. If you change the value of each probability, pie chart and probability sum will also be changed to reflect the latest information. You should make sure that sum is 1 (or very close to 1) after modifying. You can use "normalize" button to help you to achieve that. Emission probability is on the other tab of the same dialog.
 |
Modify emission probability |
You can modify emission probability for every matching, and insertion node. Emission probability for N, B, E, C, J nodes are equal to background emission frequency and can not be modified in "Hmm Model" view. However, you can modify background emission frequency by editing G node in null model. You can modify transition probability for every node except for start node S, end node T and last deletion node Dn, which has only one transition to node E.
After editing a node, a "*" mark will be appended to label of the node to indicate that it has been modified. And the thickness of connection line and node border is recaculated to reflect this change.
You can remove an existing state in pHMM. Right click a matching node and select "delete" in the pop up menu. Nodes are deleted in a group. One group, which we call one state, consists of one matching node, one deletion node and one insertion node. The number part of the node label are the same (such as D21, I21, M21). If right click "delete" on a matching node, the whole group will be deleted rather than the matching node alone. All states consist of three nodes except for last one, which only has matching and deletion node.
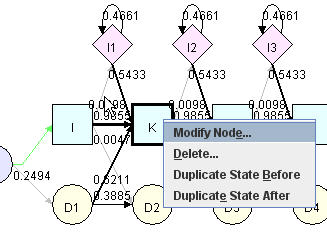 |
Node Editing Menu |
You can insert a state using the pop up menu command "Duplicate state before" or "Duplicate state after". Again, nodes are inserted in group, in our term, state. New nodes will have the transition probability and emission probability same as the selected state. However, one exception is the last state. When you duplicate last state, data of matching and deletion nodes are copied from the selected state, insertion node is copied from previous state because the last state do not have insertion node. The inserted nodes will have the same label name as the seleted state, except that a "#" mark will precede it to indicate an inserted state. The new label will not be saved in hmm file. In hmm file, only the order of states will be saved. So the first state is 1 and then 2, and so on. To see the actual state number in hmm file rather than labels reflecting deletion and insertion, you can use menu command "View"->"Renumber" to regenerate state number.
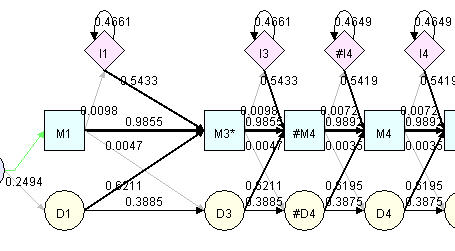 |
Inserted/Deleted/Modifed states |
In "Null Model" view, you can modify transition and emission probability as well. Bear in mind that emission probability of node G is actually background emission frequency of 20 amino acid.
Every modify/delete/insert operation in "Hmm Model"/"Null Model" view can be undo/redo using "Edit" menu.
"Hmm Logo" view and "Hmm Text" view are readonly. You can not edit anything in these two views.
Viterbi path
To align a sequence against a pHMM model, first use "File"->"Open Sequence" to open a sequence file. HMMVE support most popular sequence file formats. After open a sequence file, all sequences inside the file will be listed in sequences table in the right lower part of HMMVE.
You can double click a sequence to invoke a sequence editor. You can modify sequence in the editor. However, sequence modification can not undo using "undo" command in "Edit" menu. There is no way to undo a sequence modification in HMMVE.
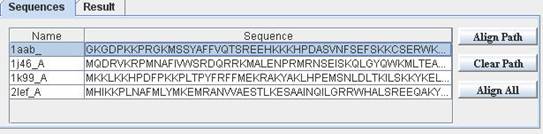 |
Sequence List |
|
Sequence Editor |
To align a sequence against a pHMM, simply select a sequence in sequences table, click "Align Path" button, you can see Viterbi path in "Hmm Model" view.
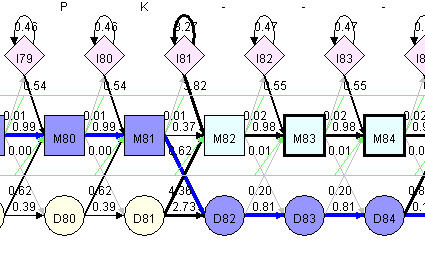 |
Viterbi Path |
When we apply pHMM to align multiple sequences, no multi-domain match (domain duplication) will be considered. So before aligning a sequence to pHMM, transition probability from E to J will be set to 0 to prevent a loop back. Multi-domain match should be valid in sequence search, however, we do not include multi-domain Viterbi path in current version of HMMVE.
After sequence alignment, blue thick line in "Hmm Model" view is the Viterbi path. On top of the model, you can see the emission of each node for that sequence. If it is a matching node, one amino acid will be emitted. This amino acid has a good chance, but is not necessarily equal to the matching node consensus. If the path go through a deletion node, no amino acid will be emitted, however, a "-" will be shown on top of model indicate there is a deleted position. If it is an insertion node, one or several inserted amino acid will be emitted. The inserted amino acid will be shown on top of model inside a parenthesis to indicate these are inserted. Except for matching, insertion and deletion node, N and C nodes also emit amino acid.
You can click "Align all" button to align all sequences in the sequence file just like "hmmalign" do. The result will be shown in "Align result" pane and you can save it into a sequence file. Only "msf" sequence file format is available to save alignment result in current version.
|
|
|
|